DNA Extraction Protocol for Adult Corals
This protocol is based on the Zymo Quick-DNA™ Miniprep Plus Kit protocol and the Putnam Lab protocol using the Zymo Duet RNA/DNA Extraction Kit written by E. Strand.
![]()
Reagent Preparation
Check kit contents and instructions to confirm prep steps.
- If buying g-DNA Wash Buffer Separately, add 96 mL 100% ethanol (104 mL 95% ethanol) to the 24 mL DNA/RNA Wash Buffer concentrate before use. The g-DNA Wash Buffer included with d4069 (Miniprep Plus Kit) is supplied ready-to-use and does not require the addition of ethanol prior to use.
- Reconstitute the lyophilized (freeze-dried) 20 mg Proteinase K with 1,040 uL Proteinase K Storage Buffer or the 5 mg lyophilized (freeze-dried) Proteinase K with 260 uL Proteinase K Storage Buffer. Vortex to dissolve. Store at -20°C.
Adult Fragment Tissue Sample Preparation
These steps remove the coral tissue from the skeleton, leaving the skeleton pieces in the bead tube and the supernatant containing degraded tissue (and DNA/RNA).
Materials
- 70% Ethanol
- RNAse Away
- KimWipes and/or paper towels
- Tweezers
- Bone clippers
- Bead tubes: 2 mL 0.5 mm glass beads and tubes from Fisher Scientific
- DNA/RNA Shield
- 1.5 mL Eppendorf tubes
Protocol
1. Sterilizing the working area
- Rinse clippers and tweezers with:
- 70% Ethanol, then wipe with Kimwipe - RNAse away, then wipe with Kimwipe - Spray gloves with 70% Ethanol and rub hands together. Then spray gloves with RNAse away and rub hands together.
- Spray bench with 70% Ethanol and RNAse away and wipe with paper towel.
Note: Take fragments one at a time out of the freezer (or store in cooler on dry ice) and sterilize the working area in between every fragment. These steps are time-sensitive to prevent the coral fragments and freezer from thawing. Do not rush, but be efficient.
2. Protocol for a whole snap-frozen fragment tissue homogenization
- Label the cap of the bead tube with the sample ID, and label the side of a 1.5 mL Eppendorf tube with the sample ID, your initials, and today’s date. Label the cap of the 1.5 mL Eppendorf tube with the coral ID number.*
- Add 1000 μl of RNA/DNA shield to the tube. *
- Remove desired fragment from -80°C freezer or cooler.
- Using sterilized clippers, clip off 3-4 small pieces and place all the pieces into the 1.5 mL microcentrifuge tube. Depending on desired DNA RNA quantity, you can increase the amount of uL added of the original DNA RNA shield from the original sampling tube or increase the size of fragment used. Note that RNA/DNA shield needs to cover the fragments.
- Vortex the 2 mL 0.5 mm glass bead tube for 1 minute for Pocillopora spp. and 2 minutes for Montipora spp.. The amount of time for vortexing will depend on the coral skeletal structure and how easily the tissue separates from the skeleton. Too much vortexing can cause DNA/RNA degradation, but too little vortexing can result in minimal DNA/RNA yield.
- After vortexing, check to see if most of the tissue has come off of the skeleton and that the DNA/RNA shield has darkened in color.
- Remove supernatant (AKA homogenate; ~1 mL) and place in a new labelled 1.5 mL microcentrifuge tube.
- Proceed to extraction steps.
*Can do steps 1& 2 in advance
DNA Extraction
Materials
In the Zymo Quick-DNA™ Miniprep Plus Kit:
- Proteinase K
- Yellow DNA spin columns and collection tubes, labelled appropriately
- Genomic Binding Buffer
- DNA Pre-Wash Buffer
- g-DNA Wash Buffer
- 10mM Tris HCl pH. 8.0
Must be supplied separately:
- 1.5 mL Eppendorf tubes
- Elution liquids (10mM Tris HCl pH. 8.0 or DNase/RNase free water)
Protocol
1. Protein digestion
- Aliquot 400 uL of the homogenate (from Step 7, Protocol for a whole snap-frozen fragment tissue homogenization) into a new 1.5 mL microcentrifuge tube for extractions. Label the cap with the fragment’s sample ID. Save the leftover aliquoted supernatant tube in -80°C freezer as a potential back-up in case the extraction doesn’t work.
- Add 20 µl of Proteinase K to each sample tube.
- Vortex for 10-15 seconds and spin down.
- Incubate the sample tubes for 15 min at room temperature.
2. DNA Binding
- Set up yellow DNA spin columns and collection tubes, label appropriately.
- Warm elution liquids to 60-70°C (10mM Tris HCl pH. 8.0 and RNase free water).
- Add equal volume (420 µl) Genomic Binding Buffer to each sample tube.
- Vortex for 10-15 seconds.
- Centrifuge at 14,000 rpm for 2-5 minutes to pellet unlysed tissues.
- Transfer the supernatent (~840 µL total volume) to a yellow DNA spin column.
- Centrifuge at 14,000 rpm for 1 minute.
- Discard the collection tube with the flow through and transfer the spin column to a new collection tube.
3. Removal of contaminants
- Add 400 µl DNA Pre-Wash Buffer gently to the yellow DNA spin columns.
- Centrifuge at 14,000 rpm for 1 minute.
- Discard flow through (Zymo kit waste).
- Add 700 µl g-DNA Wash Buffer gently to the yellow DNA spin columns.
- Centrifuge at 14,000 rpm for 1 minute.
- Discard flow through (Zymo kit waste)
- Add 200 µl g-DNA Wash Buffer gently to the yellow DNA spin columns.
- Centrifuge at 14,000 rpm for 1 minute.
- Discard flow through (Zymo kit waste).
- Centrifuge at 14,000 rpm for 1 minute to evaporate any remaining Wash Buffer on the column.
- Discard the collection tube with the flow through and transfer the spin column to a new 1.5 mL Eppendorf tube. Be careful not the get the spin column wet.
4. Elution
- Add 50 µl warmed elution liquid to each yellow DNA column by dripping slowly directly on the membrane, getting as close to the membrane as possible without touching it.
- Incubate at room temperature for 5 minutes.
- Centrifuge at 14,000 rpm for 1 minute.
- Add 50 µl warmed elution liquid to each yellow DNA column by dripping slowly directly on the membrane, getting as close to the membrane as possible without touching it.
- Incubate at room temperature for 3 minutes.
- Centrifuge at 14,000 rpm for 1 minute.
- Label tubes, store at 4°C if quantifying the same day or the next, if waiting longer store in -20°C.
Extraction Content Analysis
These steps analyze the quantity and quality of the DNA extracted and may be done on a separate day from the extraction.
DNA Quantity
Follow Broad Range or High Sensitvity dsDNA Qubit protocol to analyze sample quantity. Read all samples twice and take the average.
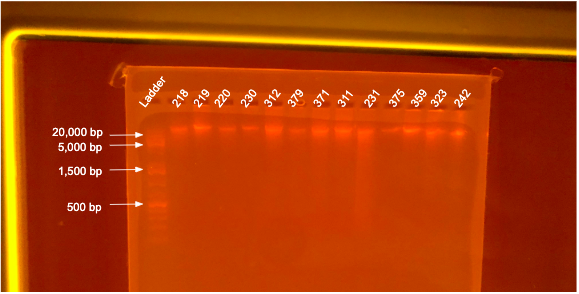
DNA Quality
If DNA quantity is sufficient (typically >10 ng/µL) follow the Bhattachrya Agarose Gel Protocol to determine DNA quality. “Good” DNA should form a distinct band a the very top of the gel. See example below: