
Mapping GO and Kegg terms to the M. capitata genome
A pipeline using DIAMOND Blast search, InterProScan, Blast2GO, UniProt, & KofamScan
Functional annotation of a Monitpora capitata reference genome
Functional annotation tags putative genes in a reference genome or transcriptome with the known functions of homologous genes in other organisms. Homologous sequences are first found using the program BLAST, which searches the reference sequence against a database of reviewed protein sequences. Once homologous sequences are found, genes are tagged with known Gene Ontology or Kegg Pathway terms for the homologous sequences. Annotation allows us to better understand the biological processes that are linked to genes of interest.
Necessary Programs and Equipment
- A high performace computer (i.e. the URI bluewaves server; Instructions to obtain server access here) with the following programs:
- DIAMOND Search Program v2.0.0
- InterProScan v.5.46-81.0
- Requires
- Java v11.0.2
- Requires
- KofamScan v1.3.0
- Requires
- Ruby v2.7.1
- parallel
- util-linux v2.34
- HMMER v3.3.1
- Requires
- A computer/laptop with with the following programs:
- Blast2GO Basic GUI v5.2.5
- R v4.0.2
- RStudio v1.3.959
Step 1: Find homologous sequences:
i) Download/Update nr database
The nr, or non-redundant, database is a comprehensive collection of protein sequences that is compiled by the National Center for Biotechnology Information (NCBI). It contains non-identical sequences from GenBank CDS translations, PDB, Swiss-Prot, PIR, and PRF, and is updated on a daily basis.
If you are in the Putnam Lab and on bluewaves
Go to the sbatch_executables subdirectory in the Putnam Lab shared folder and run the script, make_diamond_nr_db.sh. This script, created by Erin Chille on August 6, 2020, downloads the most recent nr database in FASTA format from NCBI and uses it to make a Diamond-formatted nr database.
If you are not in the Putnam Lab
Download the nr database from NCBI. Then, use Diamond’s makedb command to format the database in a Diamond-friendly format. You can also use the command dbinfo to find version information for the database.
wget ftp://ftp.ncbi.nlm.nih.gov/blast/db/FASTA/nr.gz #download nr database in fasta format
diamond makedb --in nr.gz -d nr
diamond dbinfo -d nr.dmnd
ii) Run DIAMOND Search
With your updated nr database, you can run DIAMOND. DIAMOND, like NCBI’s BLAST tool, is a sequence aligner for protein and translated DNA searches. The tool is optimized for large datasets (>1 million sequences), and is 100x-20,000x faster BLAST without losing sensitivity.
The commands that I used are below, however, the script I used to execute this on bluewaves is available on my project repository. As input, DIAMOND requires your reference sequences (either protein or CDS nucleotides), and a path to your nr database. Below, I output the results to DIAMOND format and then convert to XML. I highly suggest the DIAMOND export format, as it can easily be converted into any other format that you may need for your analysis.
It may take a few days depending on the number of sequences you have the M. cap genome (~63,000 genes) took 4.5 days
Blastx: Align translated DNA query sequences against a protein reference database
Options:
- -d - Path to nr database
- -q - Path to reference fasta file
- -o - Base output name
- -f - Output format. 100=DIAMOND output.
- -b - Block size in billions of sequence letters to be processed at a time. Larger block sizes increase the use of memory and temporary disk space, but also improve performance. Set at 20. 20 is the highest recommended value. CPU is about 6x this number (in GB).
- –more-sensitive - slightly more sensitive than the –sensitive mode.
- -e - Maximum expected value to report an alignment. 1E-05 is the cut-off typically used for sequence alignments.
- -k - Maximum top sequences. Set at 1 because I only wanted the top sequence reported for each gene.
- –unal - Report unaligned queries (yes=1, no=0).
View: Generate formatted output from DAA files
Options:
- -a - Path to input file
- -o - Base output name
- -f - Output format. 5=XML output.
#Run sequence alignment against the nr database
diamond blastx -d /data/putnamlab/shared/databases/nr.dmnd -q ../data/ref/Mcap.mRNA.fa -o Mcap.annot.200806 -f 100 -b 20 --more-sensitive -e 0.00001 -k1 --unal=1
#Converting format to XML format for BLAST2GO
diamond view -a Mcap.annot.200806.daa -o Mcap.annot.200806.xml -f 5
Step 2: Map Gene ontology terms to genome
Can be done concurrently with Steps 1 and 3
i) InterProScan
InterProScan searches the database InterPro database that compiles information about proteins’ function multiple other resources. I used it to map Kegg and GO terms to my Mcap reference protein sequences.
The commands that I used are below, however, the script I used to execute this on bluewaves is available on my project repository. As input, InterProScan requires reference protein sequences. Below, I output the results to XML format, as it is the most data-rich output file and can be used as input into Blast2GO.
Note: Many fasta files will use an asterisk to denote a STOP codon. InterProScan does not accept special characters within the sequences, so I removed them prior to running the program using the code below:
cp Mcap.protein.fa ./Mcap.IPSprotein.fa
sed -i 's/*//g' Mcap.IPSprotein.fa
Interproscan.sh: Executes the InterProScan Program
Options:
- -version - displays version number
- -f - output format
- -i - the input data
- -b - the output file base
- -iprlookup - enables mapping
- -goterms - map GO Terms
- -pa - enables Kegg term mapping
interproscan.sh -version
interproscan.sh -f XML -i ../data/ref/Mcap.IPSprotein.fa -b ./Mcap.interpro.200824 -iprlookup -goterms -pa
ii) Blast2GO
Blast2GO is a powerful annotation tool that takes the output from BLAST, and matches the IDs of the homologous sequences identified to gene ontology terms in the GO database. To map GO terms to our identified sequences, I used the DIAMOND output XML file as input for Blast2GO mapping. I then combined the output of Blast2GO with the XML file generated from InterProScan. Blast2GO mapping took about 16 hours to complete on Mcap’s 63,227 Blasted sequences and about 45 minutes for Pacuta’s Blasted 4,760 sequences.
First, open Blast2GO. Click the down arrow next to “Start” and select “Load BLAST XML”.
Once opened, your screen should look something like this:
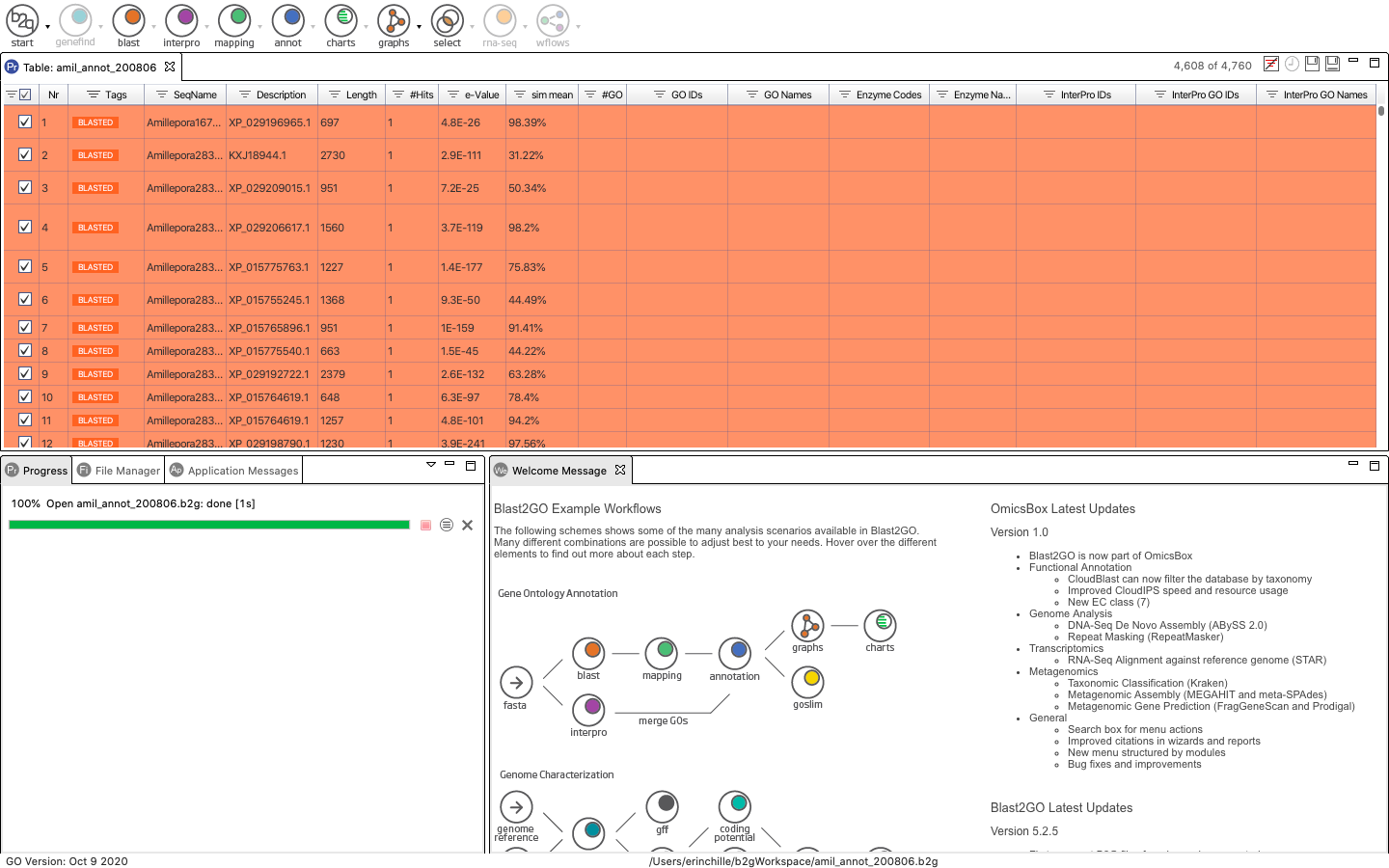
Next, click mapping to open a pop-up window where you can specify run parameters. I used all default options. Note that the below photo was taken after running all of these steps so the example sequence appears blue and includes the blast, InterProScan and Annotated Tags. Your sequences should still appear Orange.
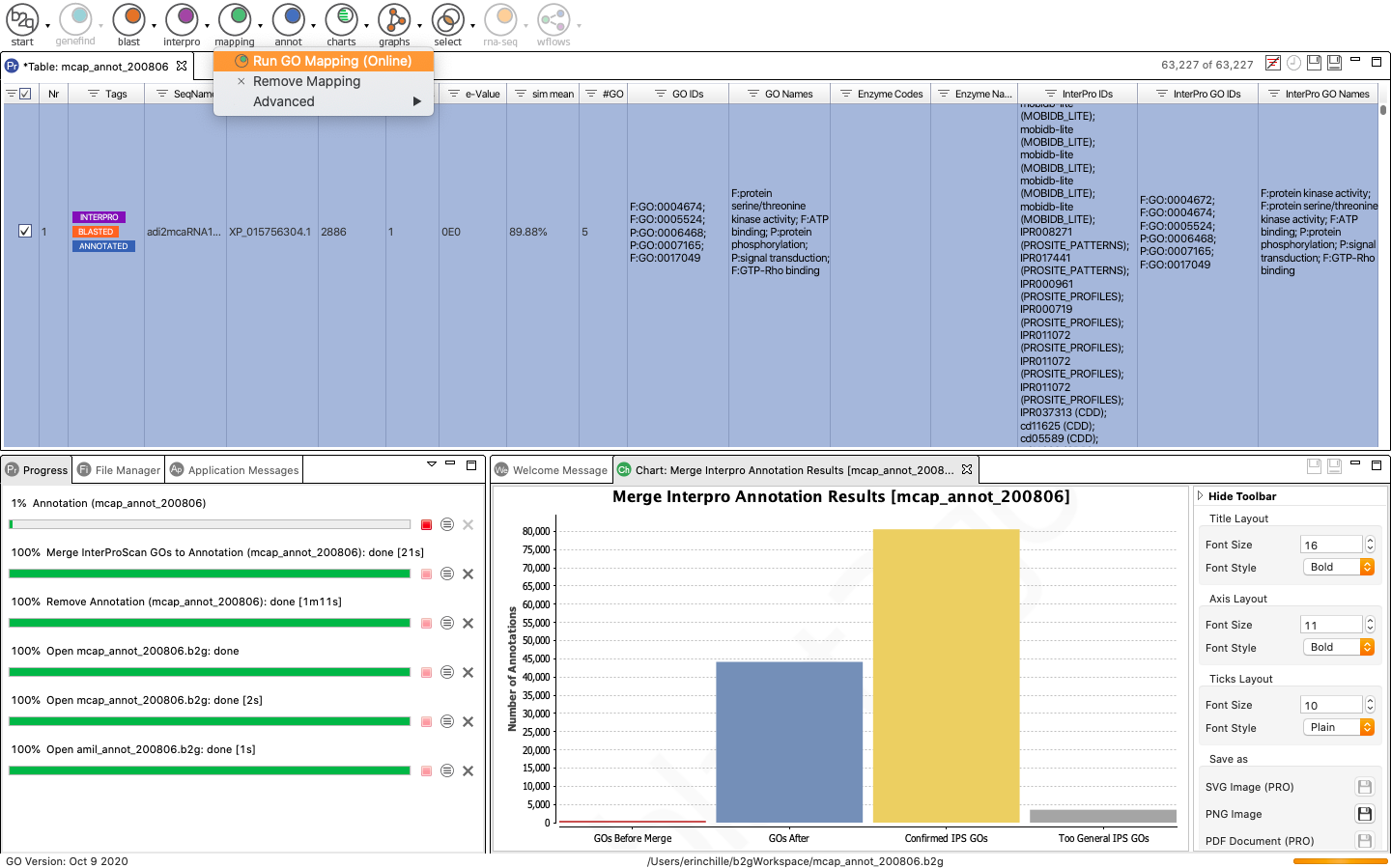
Once Blast is finished running, you can see the number of sequences that returned GO terms on the top right corner. The sequences with mapped GO terms generally appear at the top of the table while un-mapped sequences are towards the bottom. Finally, you can annotate your sequences from the GO terms obtained via the mapping step using the Annotation tool.
Now we can use Blast2GO to merge (add and validate) all GO terms retrieved via InterProScan to the GO mappings retrieved from Blast2GO
To merge your annotation results, navigate to “File > Load InterproScan Results”. Selecting this will open another pop-up window where you can upload your InterProScan XML file to the software. Be sure to specify that the InterProScan results are proteins.
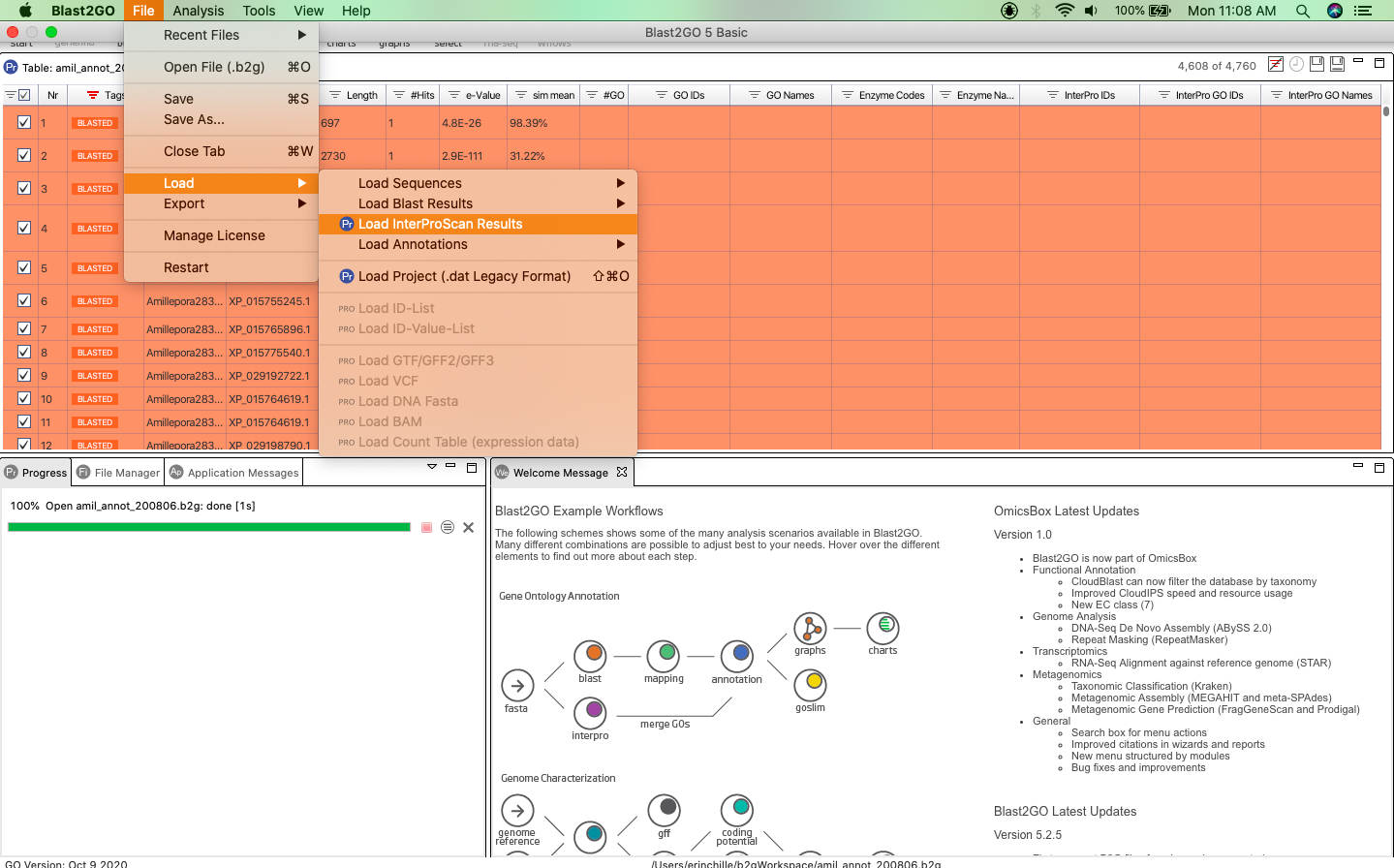
Now that your InterProScan results have been loaded, you can easily merge your Blast2GO results and InterProScan results by navigating to the Purple Interpro button and selecting “Merge InterProScan GOs to Annotation”.
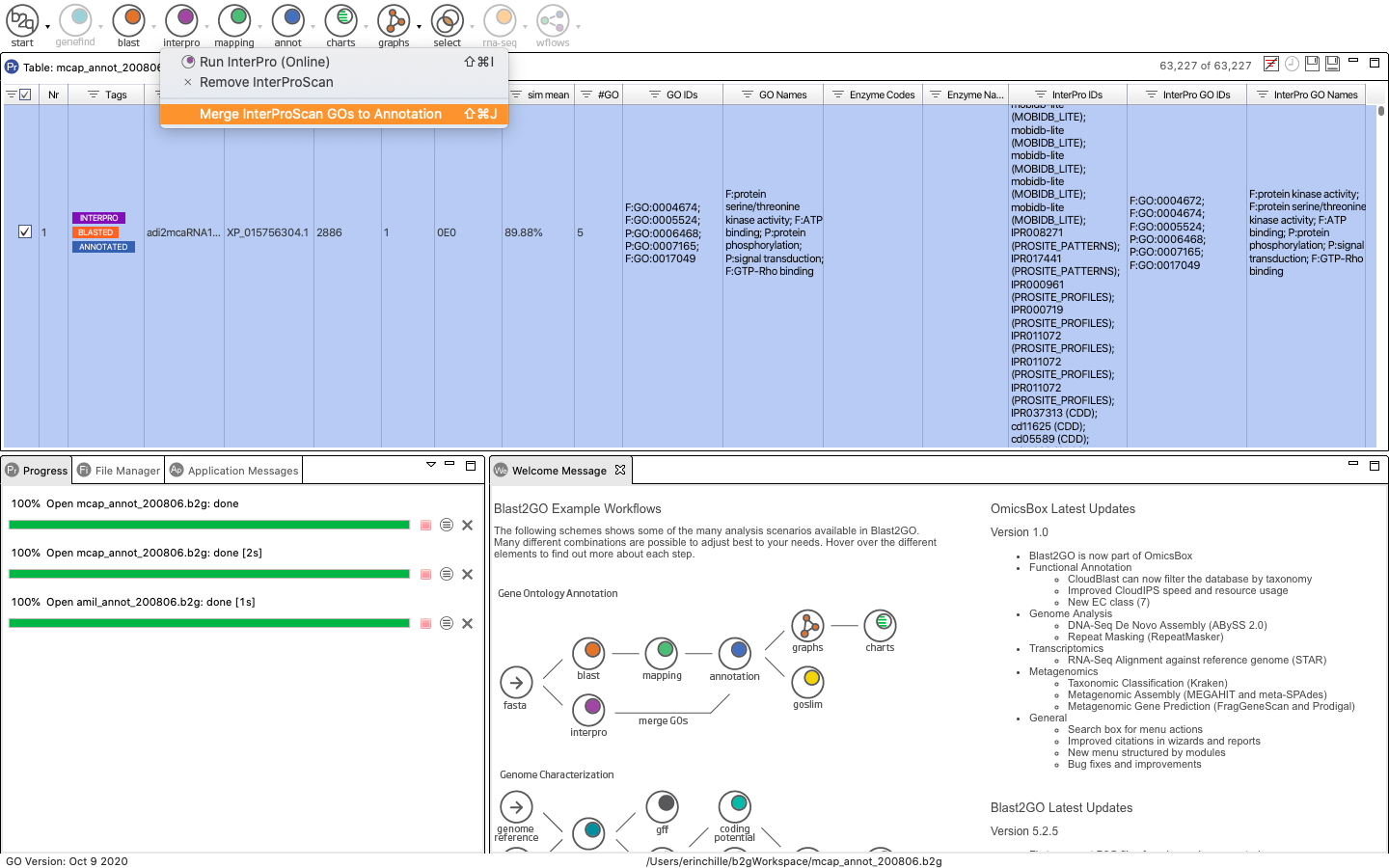
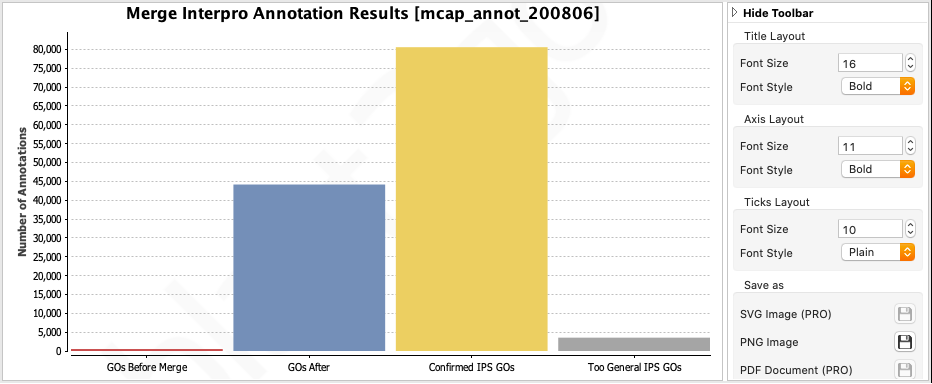
The results of the merge will appear in the bottom right window. You can see that InterProscan retrieved many more GO terms than Blast2GO (red versus blue).
iii) Uniprot
The Uniprot Retrieve ID/Mapping Tool is a useful web-based tool to gather GO and Kegg terms from the accession IDs available in it’s database. To use this tool, you need a list of accession IDs, which you can get from your Blast2GO output (The Description column). You can save this column as a separate text file to use as input.
a) Generate a list of accession IDs
To generate a list of accession IDs, I opened the output of Blast2GO in Excel and deleted the first two columns, then saved and closed the file. Next, I opened the command-line and used cat and awk to write the second column (the description column) to a new text file.
cat 1-BLAST-GO-KO/3-BLAST2GO/Mcap_blast_200806_GO_200811_Interpro_200824.txt | awk '{ print $2 }' > 1-BLAST-GO-KO/4-Uniprot/Mcap_accessions.txt
b) Upload the list to Uniprot to retrieve all hits
Uniprot has a very helpful guide to using this tool on their website. See https://www.uniprot.org/help/uploadlists. It will output a table of hits looking something like this:
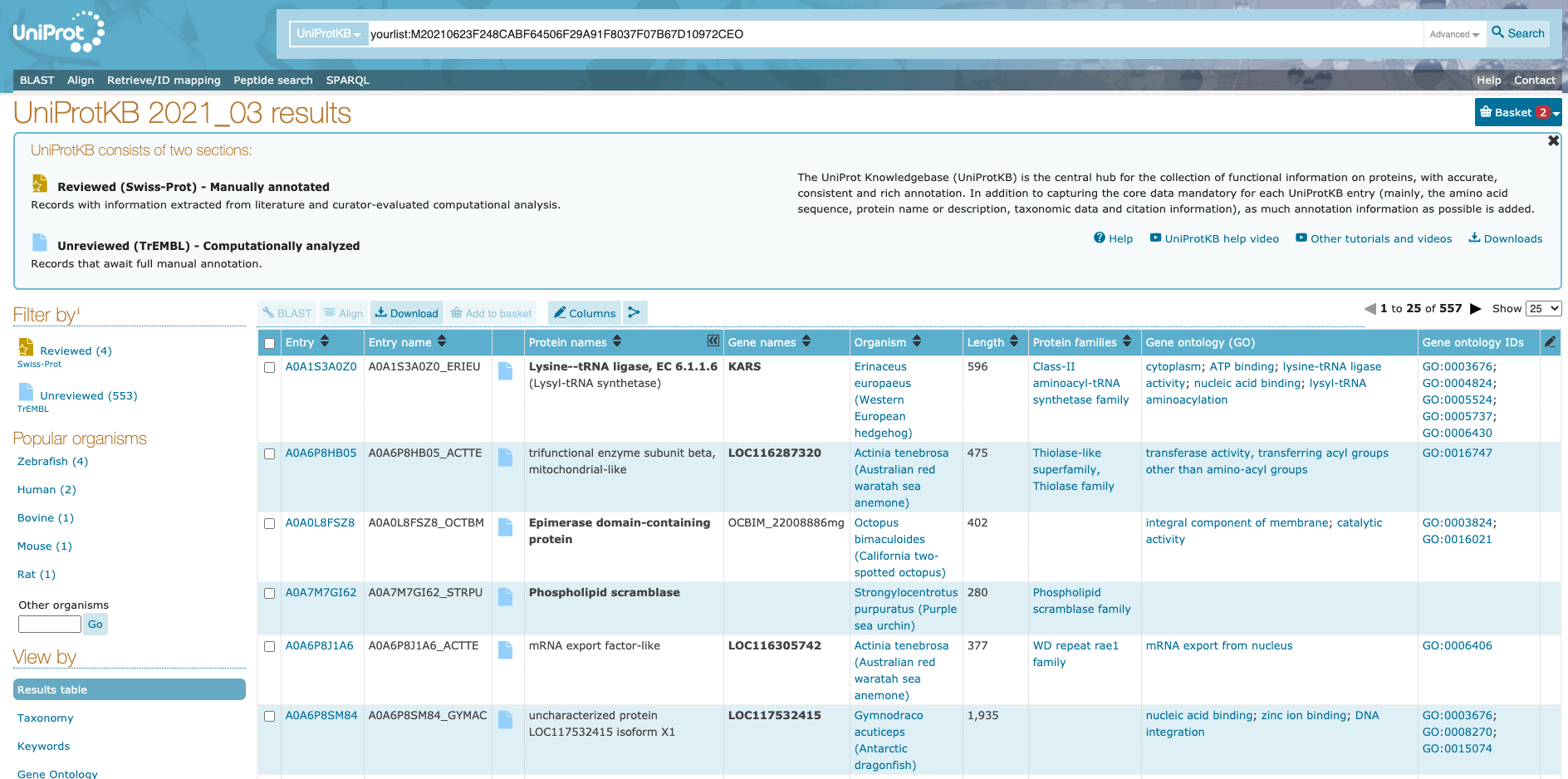
To edit the columns you want to view/download click the “Columns” button:
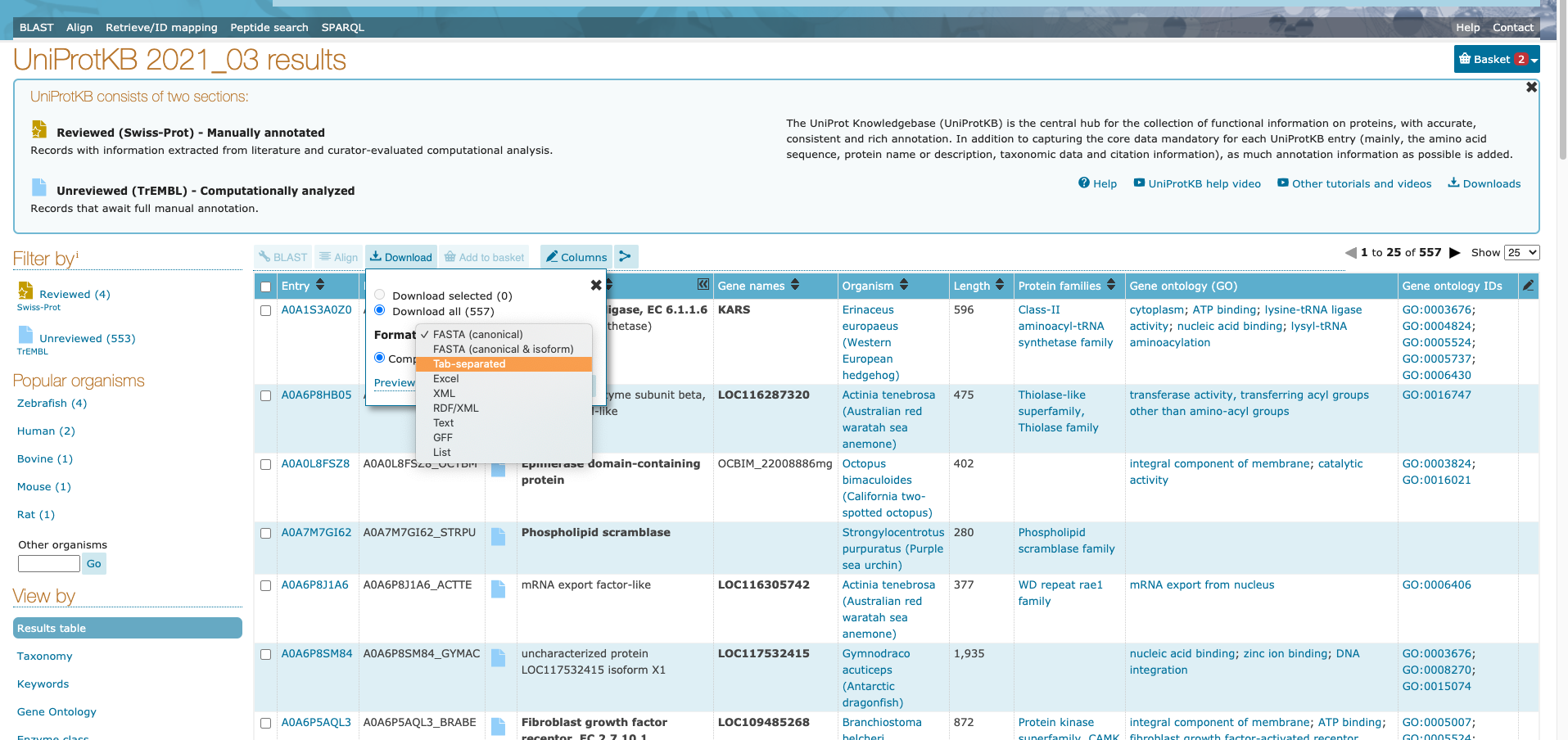
Finally, to download the table, click on the “Download” button. I downloaded mine as a tab-separated file, but GFF format may also be helpful.
Step 3: Map Kegg terms to genome
KofamScan is the command-line version of the popular KofamKOALA web-based tool, used to map Kegg terms (containing pathway information) to a genes. KofamScan and KofamKoala work by using HMMER/HMMSEARCH to search against KOfam (a customized HMM database of KEGG Orthologs (KOs). Mappings are considered robust because each Kegg term has an individual pre-defined threshold that a score has to exceed in order to map to a gene. While all mappings are outputted, high scoring (significant) assignments are highlighted with an asterisk.
The commands that I used are below, however, the script I used to execute this on bluewaves is available on my project repository. In order to run KofamScan, you will need a fasta file of predicted protein sequences (preferably the same one used to run InterProScan).
i) Download and inflate the Kofam database.
To get the most up-to-date Kofam database, download it just before running KofamScan. You will also need to download the profiles associated with the Kofam database containing threshold information.
curl -O ftp://ftp.genome.jp/pub/db/kofam/ko_list.gz | gunzip > ko_list #download and unzip KO database
curl -O ftp://ftp.genome.jp/pub/db/kofam/profiles.tar.gz | tar xf > profiles #download and inflate profiles
ii) Run KofamScan
exec_annotation: executes Kofamscan
To execute Kofamscan, use the exec_annotation script provided with program download. To run, use: exec_annotation {options} path_to_query_file.
Options (See KofamScan repo for more options):
- -o - set name of output file
- -k - path to ko database (downloaded above)
- -p - path to profile database (downloaded above)
- -E - set minimum expect value for significant mappings
- -f - output format
- –report-unannotated - returns names of sequences with no mapped KO terms
- -pa - enables Kegg term mapping
/opt/software/kofam_scan/1.3.0-foss-2019b/exec_annotation -o Mcap_KO_annot.txt -k ./ko_list -p ./profiles/eukaryote.hal -E 0.00001 -f detail-tsv --report-unannotated /data/putnamlab/erin_chille/mcap2019/data/ref/Mcap.protein.fa
Step 4: Compilation of the output of different methods
Done in RStudio. See RMarkdown page.