Workflow to test if canditate amplicon regions can reconstitute the full phylogeny with all SNPs
The following protocol describes the process that I used to create a phylogeny of our P. acuta samples from a candidate amplicon region, using the profilin region as an example.
1. Extract region of interest from full SNP vcf and export as a fasta file
Input: As input, we require 1) the full VCF file of all SNPs called during the genotyping process (GVCFall_SNPs.vcf.gz), and 2) a BED file containing the region of interest (amplicon_regions.bed).
++BED file example:++
| CHROM | START | END | ANNOTATION |
|---|---|---|---|
| Pocillopora_acuta_HIv1__Scaffold_000107F__length_1282968 | 463903 | 464598 | profilin |
Code: All code was performed on the coral Rutgers server in the Triploid-Pacu-Gene-Dosage/primer_pcr/amplicon_phylogeny directory. I used tabix, a tool in the samtools package, to extract the region of interest and used the vcf2phylip python script to export it to fasta format.
- Activate the samtools/bcftool conda environment, which uses samtools v1.15.
conda activate sam_bcf_toolpak #samtools-v1.15, #bcftools-v1.15 - Index the VCF file containing all SNPs with tabix.
tabix -p vcf input_files/GVCFall_SNPs.vcf.gz #index the vcf - Use tabix to extract the SNPs in the profilin protein. Takes as input the BED file described above (
--regions) and the full SNP VCF file. I used the flag--print-headerto keep all information in the file, most importantly, the sample names.tabix --print-header --regions input_files/amplicon_region_profilin.bed input_files/GVCFall_SNPs.vcf.gz > output_files/Pocillopora_acuta_HIv1___Scaffold_000107F___length_1282968.463903-464598.profilin.vcf - Use the vcf2phylip script to export the profilin SNP vcf file to
./scripts/vcf2phylip.py --input output_files/Pocillopora_acuta_HIv1___Scaffold_000107F___length_1282968.463903-464598.profilin.vcf --output-folder output_files --phylip-disable --fasta --write-used-sites --outgroup Pacuta_ATAC_TP7_1445 #export vcf to fasta for phylogeny - Deactive the conda environment.
conda deactivate
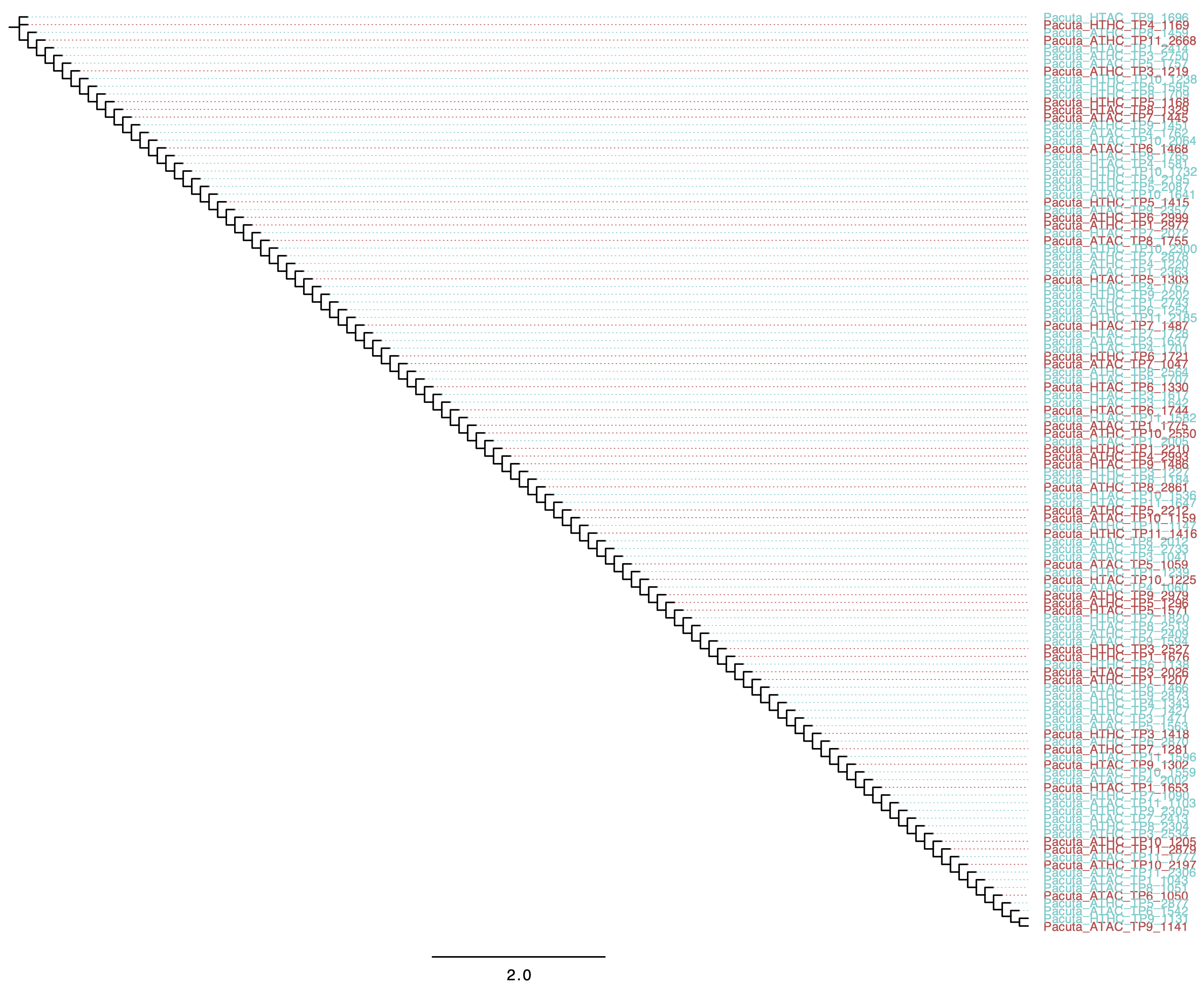
We will use Pocillopora_acuta_HIv1__Scaffold_000107F__length_1282968.463903-464598.profilin.min4.fasta as input into raxmlGUI
2) Make phylogeny with raxmlGUI (v2.0.7) and FigTree (v1.4.4) to verify region reconstitutes our full SNP phylogeny
-
Exit the Rutgers coral server and
scpthe fasta file, Pocillopora_acuta_HIv1__Scaffold_000107F__length_1282968.463903-464598.profilin.min4.fasta, to your local computer. -
Open raxmlGUI
-
Load alignment file (Pocillopora_acuta_HIv1__Scaffold_000107F__length_1282968.463903-464598.profilin.min4.fasta)
-
Ensure that the GTR model (for nucleotides) is selected
-
Run ML Tree Inference with 500 Runs
-
Select an outgroup. I selected Pacuta_ATAC_TP7_1445 because it was the weird triploid that reverted back to the diploid state.
-
Click “Run Model Test”
-
Exit RAxML and open FigTree
-
Reroot the tree by preference
-
Import clade and ploidy .txt annotation file
-
Color tree labels by clade/ploidy and export results as a pdf
Resulting Tree: